Sequencing Coverage
Introduction to Sequencing Coverage
Sequencing coverage describes the average number of reads that align to, or "cover," known reference bases. The next-generation sequencing (NGS) coverage level often determines whether variant discovery can be made with a certain degree of confidence at particular base positions.
Sequencing coverage requirements vary by application, as noted below. At higher levels of coverage, each base is covered by a greater number of aligned sequence reads, so base calls can be made with a higher degree of confidence.
Sequencing Coverage Requirements Depend on Application
Researchers typically determine the necessary NGS coverage level based on their application, as well as other factors such as reference genome size, gene expression levels, published literature, and best practices from the scientific community. Examples of sequencing coverage recommendations for some common applications are listed here.
| Sequencing Application | Recommended Coverage |
|---|---|
| Whole human genome sequencing to detect SNVs and rearrangements | 10× to 30× (depending on application and statistical model) |
| Whole-exome sequencing | 100× |
| RNA sequencing | Usually calculated in terms of numbers of millions of reads to be sampled. Detecting rarely expressed genes often requires an increase in the depth of coverage. |
| ChIP-Seq | 100× |
How to Estimate and Achieve Your Desired NGS Coverage Level
- Estimating Sequencing Coverage (PDF): Learn how to estimate the depth of coverage needed for your experiment, and read more detailed background information about sequencing coverage.
- Sequencing Coverage Calculator: Find out how to calculate the reagents and sequencing runs needed to achieve the desired sequencing coverage for your experiment.
Histograms to Depict NGS Coverage Range and Uniformity
Coverage histograms are commonly used to depict the range and uniformity of sequencing coverage for an entire data set. They illustrate the overall coverage distribution by displaying the number of reference bases that are covered by mapped sequencing reads at various depths. Mapped read depth refers to the total number of bases sequenced and aligned at a given reference base position (note that "mapped" and "aligned" are used interchangeably in the sequencing community).
In a sequencing coverage histogram, the read depths are binned and displayed on the x-axis, while the total numbers of reference bases that occupy each read depth bin are displayed on the y-axis. These can also be written as percentages of reference bases.
Examples of Good and Poor Coverage Histograms
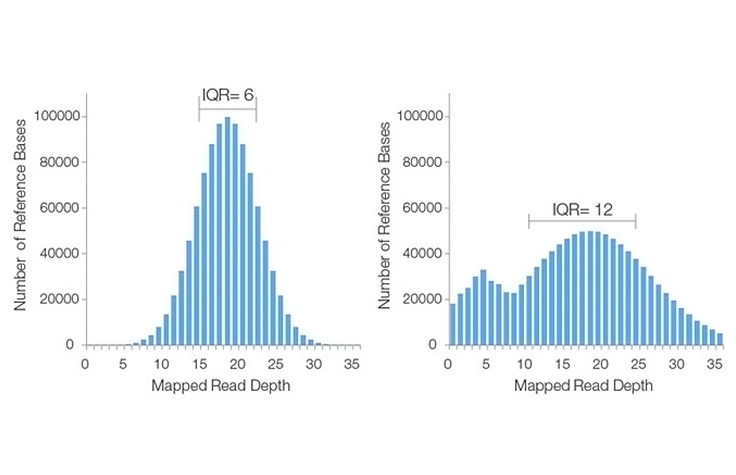
Ideally, the plot will take the form of a Poisson-like distribution with a small standard deviation, as seen in the left-hand histogram image. This distribution is valid under the assumption that reads are randomly distributed across the genome and that the ability to detect true overlaps between reads is constant within a sequencing run. However, for a variety of reasons, actual coverage histograms may have a large spread (i.e., broad range of read depths), or have a non-Poisson distribution, as seen in the right-hand histogram image.
Evaluating Next-Generation Sequencing Coverage
The following metrics are commonly used to evaluate NGS coverage:
Inter-Quartile Range (IQR)
The IQR is the difference in sequencing coverage between the 75th and 25th percentiles of the histogram. This value is a measure of statistical variability, reflecting the non-uniformity of coverage across the entire data set. A high IQR indicates high variation in coverage across the genome, while a low IQR reflects more uniform sequence coverage. In the histograms above, the lower IQR indicates that the histogram on the left has better sequencing coverage uniformity than that on the right.
Mean (Mapped) Read Depth
The mean mapped read depth (or mean read depth) is the sum of the mapped read depths at each reference base position, divided by the number of known bases in the reference. The mean read depth metric indicates how many reads, on average, are likely to be aligned at a given reference base position.
Raw Read Depth
This is the total amount of sequence data produced by the instrument (pre-alignment), divided by the reference genome size. Although raw read depth is often provided by sequencing instrument vendors as a specification, it does not take into account the efficiency of the alignment process. If a large fraction of the raw sequencing reads are discarded during the alignment process, the post-alignment mapped read depth can be significantly smaller than the raw read depth.